





Reconozca los síntomas, identifique la enfermedad
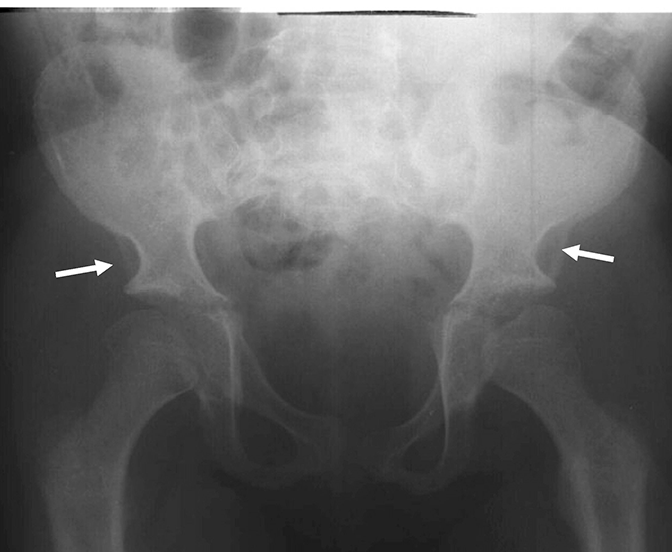
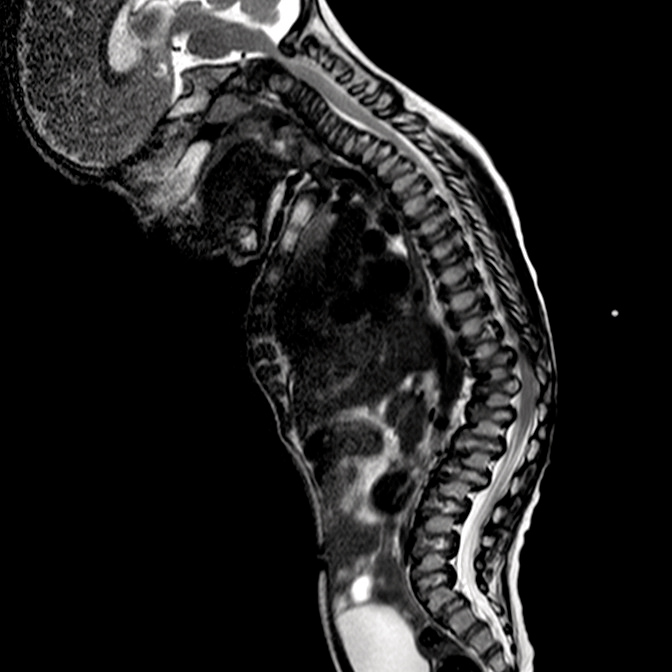
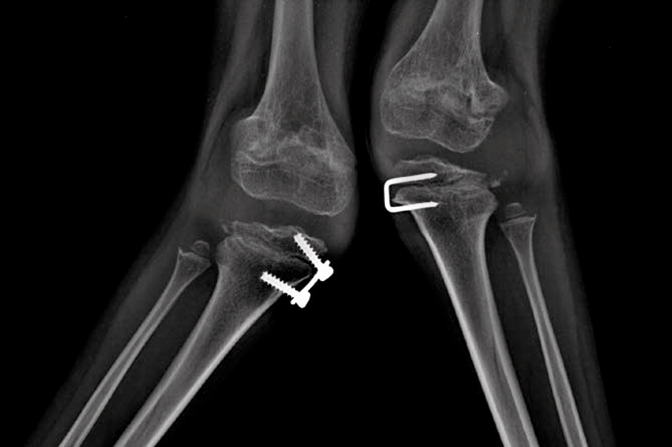
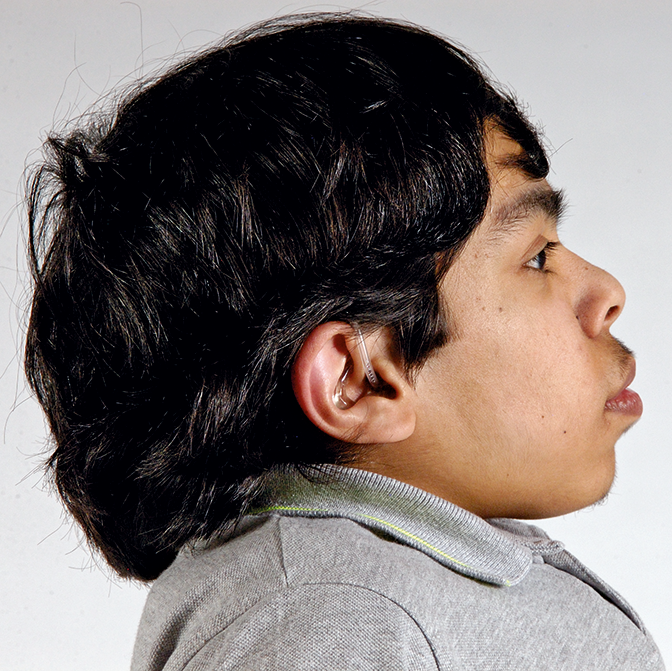
Las mucopolisacoridosis (MPS) son un grupo de enfermedades progresivas, de transmisión genética, serias, que afectan múltiples sistemas y funciones del organismo como resultado de deficiencias enzimáticas de expresión heterogénea.1-3 Las distintas manifestaciones y grados de progresión de la enfermedad —sumados a la poca frecuencia con la que aparecen en la práctica clínica — resultan en la falta de identificación de las MPS en las consultas médicas, lo que genera un diagnóstico tardío y consecuencias potencialmente devastadoras.4,5
Optimice los resultados de los pacientes a largo plazo y colabore con el inicio de un tratamiento temprano, identificando los signos de las MPS y derivando los pacientes con sospecha de la enfermedad; a un genetista o centro del metabolismo de inmediato. La consulta a un centro del metabolismo es un primer paso fundamental para el diagnóstico diferencial y un pronto inicio de tratamiento.4
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}